Image courtesy of Toshiba Medical.
April 14, 2017 ― Researchers from the University of Missouri School of Medicine have developed an imaging technique using a specific form of helium to measure the effectiveness of a new drug for cystic fibrosis. They hope the finding could lead to improved therapies for cystic fibrosis and other lung conditions.
According to the Cystic Fibrosis Foundation, more than 30,000 Americans are living with the disorder. It currently has no cure, though the new drug, approved by the U.S. Food and Drug Administration (FDA), treats the underlying cause of the disease. However, the drug’s effectiveness for each individual is unknown.
“People with cystic fibrosis have an imbalance of salt in their bodies caused by the defective CFTR protein,” said Talissa Altes, M.D., chair of the Department of Radiology at the MU School of Medicine and lead author of the study. “The drug ivacaftor targets this defective protein, but to what extent it is successful is not well understood. Our study sought to use a new way of imaging the lung to understand how well the drug is working in patients with a specific gene mutation known as G551D-CFTR.”
Cystic fibrosis causes the buildup of thick mucus that can clog airways and lead to dangerous infections. Although advances in the understanding and treatment of cystic fibrosis have allowed many people with the condition to live into their early 40s, many patients with cystic fibrosis die of respiratory failure.
Currently, lung function is measured using a test known as spirometry, in which patients blow through a tube; their progress is tracked over time. However, this method is not well suited for pediatric patients because it requires concentration, controlled breathing and cooperation with the spirometry technician. Computed tomography (CT) scans can be used, but they provide structural images of the lungs and do not show gas or airflow.
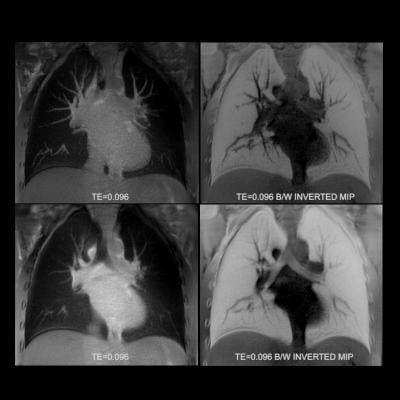
Altes and her team decided to use a specific form of helium, helium-3, as a harmless contrast agent in conjunction with magnetic resonance imaging (MRI) to visualize lung function. The study was conducted in two parts. In part one, eight patients were given either ivacaftor or a placebo for four weeks to measure short-term effectiveness. In part two, nine patients received ivacaftor for 48 weeks to determine long-term effectiveness. After each phase, patients performed a spirometry test and underwent MRI imaging using hyperpolarized helium.
“We found that after using ivacaftor, patients experienced a dramatic increase in lung improvement in both the short and long term,” Altes said. “On an MRI, a healthy lung should look completely white when helium-3 is used as a contrast agent. Conversely, areas that are not white indicate poor ventilation. That’s the beauty of this technique — it’s very obvious if the drug is working or not.”
With more study, Altes hopes to apply helium-3 MRI to younger children or babies with impaired lung function or other respiratory diseases.
“More drugs are under development to treat cystic fibrosis and other lung conditions, and improved imaging techniques are needed to test their effectiveness,” Altes said. “The importance of this technique is that it may well be a cost-effective tool to aid in the development of these drugs. However, it also can help patients know which medications may work best for their unique conditions.”
The study, “Use of Hyperpolarized Helium-3 MRI to Assess Response to Ivacaftor Treatment in Patients with Cystic Fibrosis,” recently was published in the Journal of Cystic Fibrosis. Research reported in this publication was supported by Vertex Pharmaceuticals Inc., the manufacturer of ivacaftor, the Hartwell Foundation and Siemens Healthcare.
For more information: www.cysticfibrosisjournal.com

 July 15, 2026
July 15, 2026 







